Xantogranuloma juvenil solitario cerebral
Wladimir Yanez Perez,1 José Leonardo Acosta2
1. Especialización en Neurocirugía, Colegio de Ciencias de la Salud, Universidad San Francisco de Quito, Quito, Ecuador
2. Servicio de Neurocirugía, Hospital Vozandes, Quito, Ecuador
Recibido: 22/06/2024. Aceptado: 11/10/2024
Wladimir Yanez Perez
wladimiryanez@gmail.com
DOI: 10.59156/revista.v38i04.648
ORCID
Wladimir Yanez Perez: 0000-0001-8715-8262
José Leonardo Acosta: 0000-0001-5607-7056
EMAILS
José Leonardo Acosta: jlaq2001@hotmail.com
Los autores no declaran conflicto de interés
Los autores no declaran financiamiento.
RESUMEN
Introducción: el xantogranuloma es una enfermedad benigna poco frecuente, con predominio en la población pediátrica donde existe proliferación de histiocitos, produciendo manifestaciones cutáneas y, muy rara vez, afectación en el sistema nervioso central.
Objetivo: describir las manifestaciones clínicas, hallazgos en imágenes y procedimiento quirúrgico a propósito de un caso muy raro compatible con esta patología.
Descripción del caso: paciente masculino de 8 años que debuta con crisis convulsivas secundarias a lesión tumoral única temporal izquierda con características quísticas y picos de colina en la espectroscopia por resonancia.
Intervención: craneotomía y resección tumoral microquirúrgica. Posterior estudio histopatológico en el que se evidenciaron histiocitos y células plasmáticas y positividad en inmunohistoquímica de LCA, CD138 y CD68. Se diagnosticó un xantogranuloma solitario cerebral, considerado un hallazgo raro y excepcional.
Conclusión: la planificación exhaustiva y completa previa a la intervención quirúrgica de estas lesiones cerebrales, así como el procedimiento quirúrgico diagnóstico/terapéutico y, finalmente, el estudio anatomopatológico adecuado son esenciales para garantizar los mejores resultados en pacientes con este tipo de patología.
Palabras clave: Histiocitosis. Neurocirugía pediátrica. Xantogranuloma cerebral. Xantogranuloma juvenil.
Solitary juvenile xanthogranuloma of the brain
ABSTRACT
Background: xanthogranuloma is a rare benign disease predominantly in the pediatric population, where there is proliferation of histiocytes, producing cutaneous manifestations and very rarely affectation of the central nervous system.
Objective: to describe the clinical manifestations, imaging findings and surgical procedure regarding a very rare case compatible with this pathology.
Case description: an 8-year-old male patient who debuted with seizures secondary to a single tumor lesion in the left temporal lobe with cystic characteristics and choline peaks in resonance spectroscopy.
Surgery: craniotomy and microsurgical tumor resection. Subsequent histopathological study where the presence of histiocytes and plasma cells was evident, as well as positivity in immunohistochemistry of LCA, CD138 and CD68, thus diagnosing a solitary cerebral xanthogranuloma, considered an exceptional and rare finding.
Conclusion: thorough and complete planning prior to surgical intervention for these brain lesions, as well as the diagnostic/therapeutic surgical procedure and finally the appropriate anatomopathological study are essential to ensure the best results in patients with this type of pathology.
Keywords: Histiocytosis. Pediatric neurosurgery. Cerebral xanthogranuloma. Juvenile xanthogranuloma.
INTRODUCCIÓN
El xantogranuloma es un trastorno benigno y raro en pacientes pediátricos causado por la proliferación de histiocitos. Estos histiocitos por lo general son células no Langerhans con alto contenido de lípidos que forman múltiples nódulos de color rojo amarillento con mayor frecuencia como manifestaciones cutáneas, sin embargo, existen manifestaciones sistémicas o extracutáneas como el sistema nervioso central, hígado, bazo, pulmones, riñones, ojos, entre otros. Los casos reportados de xantogranuloma solitario sin manifestaciones cutáneas son infrecuentes.1,2
Según la clasificación de histiocitosis y neoplasias de células del linaje de macrófagos-dendritas de Weitzman y Jaffe modificada, el xantogranuloma se encuentra categorizado como grupo “C” de histiocitosis cutáneas y mucocutáneas, y como proliferación de células no Langerhans (Tabla 1).3
TABLA 1. HISTIOCITOSIS DE CÉLULAS NO LANGERHANS CUTÁNEAS Y MUCOSAS
Histiocitosis de células no Langerhans cutáneas |
|
Xantogranuloma familiar |
Xantogranuloma juvenil |
Xantogranuloma adulto |
|
Reticulohistiocitoma solitario |
|
Histiocitosis cefálica benigna |
|
Histiocitosis eruptiva generalizada |
|
Histiocitosis nodular progresiva |
|
Xantogranuloma no familiar |
Enfermedad de Rosai-Dorfman |
Xantogranuloma necrobiótico |
|
Histiocitosis cutánea inespecífica |
|
Histiocitosis de células no Langerhans cutáneas con mayor componente sistémico |
|
Xantogranuloma familiar |
Xantoma diseminatum |
Xantogranuloma no familiar |
Reticulohistiocitosis multicéntrica |
Clasificación de Weitzman y Jaffe modificada del 2016.
A nivel intracraneal, son menos los casos que en órgano visceral solitario, y cuando aparece lo hace como lesión inflamatoria crónica. El xantogranuloma tiene predominio por la región selar, secundaria a hemorragia o inflamación de patología previa como quiste de bolsa de Rathke, craneofaringioma, adenoma de hipófisis o por los plexos coroideos del tercer ventrículo; sin embargo, puede aparecer en cualquier tejido intracraneal, por lo que la clínica dependerá de la localización de la lesión, entre las que destacan: crisis convulsivas, retraso mental y de crecimiento, diabetes insípida, ataxia, hemiplejia, etc.2,4
Dhener y cols. realizaron una revisión sistemática con 174 casos donde evidenciaron que el 5.74% tuvo manifestaciones extracutáneas y de estos, solo 2 tuvieron repercusión en el sistema nervioso central, lo que representa el 1.14% de todos los casos revisados.5
Objetivos
Describir y presentar un caso clínico de xantogranuloma juvenil solitario, resaltando la importancia del procedimiento quirúrgico tanto para el diagnóstico como el tratamiento.
DESCRIPCIÓN DEL CASO
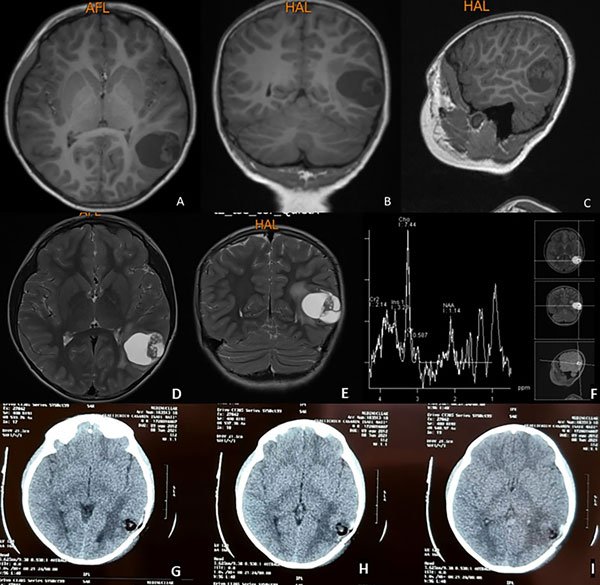
Paciente masculino de 8 años sin antecedentes patológicos conocidos, con crisis convulsivas tonicoclónicas generalizadas de 20 segundos de duración, sin presencia de aura ni relajación de esfínteres (estado postictal de 1 minuto y recuperación completa) de 2 meses de evolución. En un inicio los episodios tenían una frecuencia de 2 por semana, para alcanzar en los últimos 15 días 4 crisis diarias. A la consulta se indica levetiracetam con buena respuesta y se realiza resonancia magnética que evidencia lesión intraaxial temporal izquierda quística, hipointensa en T1 e hiperintensa en T2 y FLAIR con nódulo en su interior. La lesión mide aproximadamente 4 cm de diámetro mayor con escaso edema perilesional y moderado efecto de masa, sin herniación subfalcial y cisternas de la base visibles. La espectroscopia demostró presencia de picos de colina (Figura 1).
Figura 1. Resonancia magnética simple en secuencia T1. Se evidencia lesión ocupativa de espacio intraaxial temporal izquierda, quística, corticosubcortical hipointensa. A) Corte axial. B) Corte coronal. C) Corte sagital. En secuencias T2: D) Corte axial E) Corte coronal. F) Espectroscopía cerebral con pico de colina. G, H, I) Tomografía axial computarizada postquirúrgica: se evidencia estigma de craneotomía temporal izquierda, resección tumoral completa, lecho tumoral sin sangrado con material de hemostasia, escaso neumoencéfalo y moderado edema vasogénico.
Intervención
Se planificó tratamiento quirúrgico y se realizó craneotomía temporal izquierda, apertura dural e identificación de la lesión tumoral; se procedió a punción directa del quiste intratumoral en la que se obtuvieron, aproximadamente, 10 ml de un líquido amarillento. Mediante técnica microquirúrgica se identificó el nódulo tumoral y se hizo resección completa; no se resecó la cápsula tumoral ya que esta era muy fina y en algunos sectores era indistinguible del tejido cerebral (Figura 2).
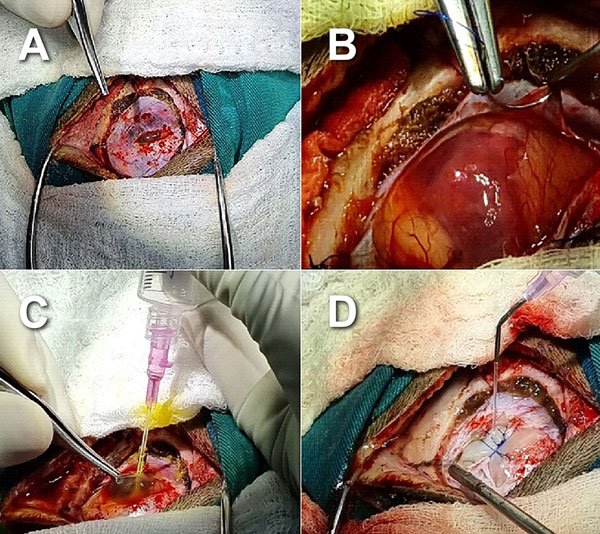
Figura 2. Imágenes transquirúrgicas. A) Durotomía. B) Aspecto quístico de lesión posterior a durotomía. C) Punción directa del quiste y obtención de líquido intratumoral amarillento. D) Duroplastia al finalizar exéresis tumoral.
Citología: líquido intratumoral con fondo proteínico con hemorragia leve. Se observan elementos celulares aparentemente histiocitarios de citoplasmas amplios vacuolados y células xantomizadas, se acompañan de células sueltas plasmáticas y linfocitos.
Histopatología: tumor intraaxial temporal compuesto por una población celular heterogénea: células de citoplasmas, amplios claros xantomizados con discreto pleomorfismo nuclear (Figura 3A), alternado con células de menor tamaño con núcleos excéntricos y nucléolos conspicuos (Figura 3B), así como linfocitos y células plasmáticas formando agrupaciones alrededor de la lesión (Figuras 3C y 3D).
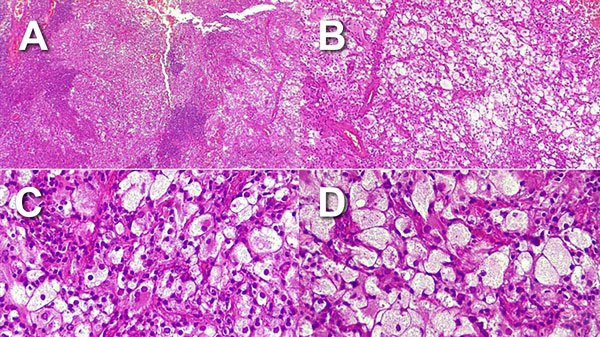
Figura 3. Histopatología con coloración de hematoxilina y eosina.
Inmunohistoquímica: reveló positividad para LCA +++/+++ en membrana de linfocitos acompañantes, positividad también para CD138 +++/+++, así como positividad para CD68 +++/+++ confirmando estirpe histiocitaria. En contraste, se demostró negatividad para S100, PGFA, sinaptofisina, citoqueratina, CD1a, IDH-1 y BRAF-V600E (Figura 4).
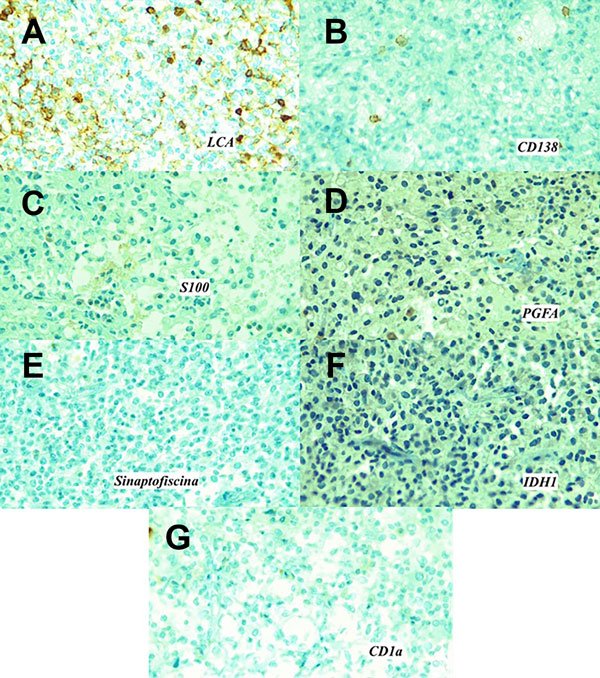
Figura 4. A) Antígeno leucocitario común (LCA / CD45). Población linfoide positiva. B) CD138 (sindecano-1) positivo. Inmunohistoquímica negativa para C) Proteína S100. D) Proteína gliofibrilar ácida (PGFA) (marcador de diferenciación glial). E) Sinaptofisina, (asociada a la vesícula presináptica). F) IDH-1 (isocitrato deshidrogenasa), no presenta mutación. G) CD1a.
DISCUSIÓN
El xantogranuloma juvenil es una entidad poco frecuente que se presenta sobre todo con manifestaciones cutáneas. Se clasifica como lesión de histiocitos de células no Langerhans. La incidencia disminuye en caso de aparecer como lesión solitaria en órganos viscerales y existen pocos casos de lesión solitaria intracraneal. La mayoría de los casos se evidencian con origen en plexos coroideos de tercer ventrículo o en región selar, lo que representa el 1% de todos los xantogranulomas.6
Se ha propuesto que las lesiones cutáneas múltiples son un factor de riesgo para el compromiso sistémico. Este hallazgo se encontró en el 67% de los pacientes con xantogranuloma sistémico, teniendo un período de latencia de meses a varios años.7 Es muy raro ver casos con lesiones solitarias sin compromiso cutáneo como en el caso expuesto.
Las manifestaciones clínicas en el sistema nervioso central tienden a ser muy variadas y dependen de la localización de la lesión: generalmente causan efectos compresivos de estructuras adyacentes o, como en el caso aquí reportado, crisis convulsivas por su localización en zonas altamente epileptogenas (lóbulo temporal).
La identificación de esta patología depende casi exclusivamente de los hallazgos histopatológicos, ya que por imágenes preoperatorias el diagnóstico diferencial es muy extenso. En nuestro caso, al tratarse de una lesión única intraaxial quística, hay que hacer diagnóstico diferencial con lesiones benignas como quistes dermoides o epidermoides, quiste aracnoideo, quistes glioependimarios o coloides, quistes de plexos coroideos o subependimarios; también lesiones infecciosas como hidatidosis o abscesos cerebrales, y lesiones neoplásicas como astrocitoma pilocítico, meningioma quístico o, inclusive, variantes anatómicas.8
Los hallazgos de espectroscopia encontrados tampoco fueron concluyentes dado el pico de colina y N-acetilaspartato (NAA). Hay que recordar que el pico de colina aumenta cuando se incrementa la síntesis de membrana celular por degradación secundaria, por ejemplo, en tumores. A su vez, el N-acetilaspartato se utiliza como marcador de número y función neuronal, es decir, todo lo que produce destrucción neuronal originará disminución de este metabolito, o en lesiones que no contengan neuronas se verá reflejada su disminución.9,10 Al realizar la comparativa de ambos metabolitos en el estudio mencionado, se evidencia una relación colina/NAA elevada, lo que puede llevar a pensar en una lesión glial de alto grado, sin embargo, en estos casos se tendría que evidenciar también un aumento de la necrosis en contexto de aumento de metabolitos como lípidos o lactato, o que, como en este caso, no sea una lesión de origen neuronal y que el pico de colina observado sea por un aumento en la síntesis de la membrana.
La inmunohistoquímica en la mayoría de estos pacientes demuestra positividad para CD68, vimentina, VIIIa, HAM56 y LYS; así como negatividad a CD1a y S100.2 En el caso expuesto, se presenta con inmunohistoquímica positiva solo para LCA, el cual es un antígeno de expresión para población linfoide, así como el CD138 y el CD68, los que tienen relación con células plasmáticas y marcador de histiocitos, respectivamente.
Es por ello que consideramos imperativo el tratamiento neuroquirúrgico (resección completa de la lesión o biopsia) para el alivio de la sintomatología y/o esclarecer la estirpe celular. Para la decisión del procedimiento quirúrgico a seguir se debe tener en cuenta: la localización de la lesión, tamaño, relación con estructuras vasculares y áreas elocuentes cerebrales, probabilidad de resección completa, riesgo de lesión cerebral y comorbilidades del paciente que van a influir directamente en los riesgos operatorios y en el desenlace clínico final.
En nuestro caso, debido al tamaño y localización de la lesión (lóbulo temporal izquierdo a nivel corticosubcortical) se decidió proceder con la resección tumoral mediante craneotomía temporal, no consideramos solamente la punción y/o biopsia del quiste ya que se contempló que el estudio histopatológico no se habría llevado a cabo de forma satisfactoria debido a la calidad de muestras que se podrían haber obtenido. Además, el riesgo de lesión de áreas elocuentes se minimizó por las características propias de la lesión y gracias a las técnicas microquirúrgicas empleadas en la resección.
CONCLUSIÓN
En el manejo de lesiones intracerebrales sin etiología clara mediante estudios de imagen o análisis clínico, la indicación quirúrgica es mandatoria, tanto para diagnóstico histopatológico como para tratamiento de los síntomas producidos por la lesión per se, garantizando la resección tumoral completa ya que esta es curativa.
En los casos en los cuales la localización de la lesión fuera inaccesible y/o irresecable, o existieran lesiones multicéntricas, la biopsia (estereotáxica o por neuronavegación) es la conducta a seguir para esclarecer el diagnóstico y seleccionar los casos en los cuales la quimioterapia coadyuvante está indicada. Es importante señalar que se debe implementar un manejo multidisciplinario de estos pacientes para garantizar el éxito en el tratamiento de esta enfermedad poco frecuente.
Contribuciones de autoría
Conceptualización: Wladimir Yanez Perez
Curación de datos: José Leonardo Acosta
Análisis formal: Wladimir Yanez Perez
Adquisición de fondos: José Leonardo Acosta
Investigación: Wladimir Yanez Perez
Metodología: José Leonardo Acosta
Administración del proyecto: Wladimir Yanez Perez
Recursos: José Leonardo Acosta
Software: Wladimir Yanez Perez
Supervisión: José Leonardo Acosta
Validación: Wladimir Yanez Perez
Visualización: José Leonardo Acosta
Redacción - borrador original: Wladimir Yanez Perez. José Leonardo Acosta
Redacción - revisión y edición: Wladimir Yanez Perez. José Leonardo Acosta
Este es un artículo de acceso abierto bajo la licencia CC BY-NC https://creativecommons.org/licenses/by-nc/4.0/
BIBLIOGRAFIA