“Este caso me desorientó”
Teyssandier, Mariano; Ángel Reyes, Keila; Recchia, Rodrigo; Melgarejo, Ana
División Neurocirugía, Hospital General de Agudos “Dr. Juan A. Fernández”, Ciudad Autónoma de Buenos Aires, Argentina
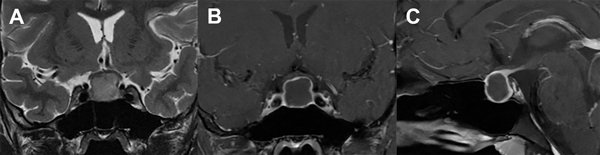
Paciente de sexo masculino de 17 años de edad, sin antecedentes de relevancia, que consultó a la guardia del hospital por presentar fiebre, cefalea intensa y vómitos de 48 horas de evolución. Al examen neurológico se encontraba lúcido, sin alteración de pares craneales, sin déficit motor ni sensitivo y presentaba rigidez de nuca. Se interpretó el cuadro como un síndrome meníngeo, por lo que se realizó un análisis de sangre que evidenció leucocitosis leve, una tomografía computada (TC) de encéfalo informada como normal y una punción lumbar. El examen físico-químico del LCR evidenció un color cristal de roca, 1425 células/mm3 con predominio mononuclear, glucorraquia de 53 mg/dl (glucemia de 102 mg/dl) y proteínas de 110 mg/dl, interpretándose como una meningitis, iniciándose tratamiento antibiótico y antiviral empírico. La serología para HIV y VDRL fueron negativas. Los cultivos de LCR fueron negativos para gérmenes comunes, micobacterias y hongos, así como también el panel viral, por lo que se interpretó como una meningitis aséptica. Se completó estudio con imagen por resonancia magnética (IRM) de encéfalo que evidenció una lesión expansiva selar con extensión supraselar de 14 x 16 mm de diámetro, quística, hipointensa en T1, levemente hiperintensa en T2, sin restricción en secuencia de difusión, con refuerzo periférico tras el contraste, con compresión y deformidad del quiasma óptico (Fig. 1). Tras este hallazgo, se realizó un campo visual computarizado que fue normal y se plantearon diferentes diagnósticos diferenciales: quiste de la bolsa de Rathke, craneofaringioma y adenoma quístico. El panel hormonal no evidenció alteración de la función hipofisaria y el paciente no refería trastornos en el crecimiento, ni en la libido, ni tampoco signosintomatología de diabetes insípida u otra alteración hidroelectrolítica. Tampoco se evidenciaron calcificaciones de la lesión en la TC. Se discutió el caso y se decidió la conducta quirúrgica.
Figura 1. A: IRM T2 coronal en la que se visualiza tumor selar con desplazamiento del quiasma óptico. B y C: IRM T1 coronal y sagital con gadolinio que muestran centro hipointenso con captación periférica en anillo.
Se realizó abordaje endoscópico transnasal transesfenoidal (ver video). Luego de la durotomía selar, se evidenció salida de material amarillento, purulento, a presión (Fig. 2). Se evacuó todo el contenido y se resecaron las paredes de la lesión. El extendido intraoperatorio fue compatible con craneofaringioma.
El paciente presentó buena evolución postoperatoria. Se realizó TC de control que no evidenció complicaciones y se le otorgó el egreso hospitalario a los 5 días postoperatorios, sin déficit neurológico ni oftalmológico.
A los 3 meses de la cirugía se realizó una IRM que evidenció una hipófisis conservada, sin alteraciones ni realces patológicos, con quiasma óptico libre y bien posicionado (Fig. 3).
La anatomía patológica diferida reveló tejido adenohipofisario con un infiltrado linfoplasmocitario crónico, compatible con una hipofisitis linfocitaria (Fig. 4).
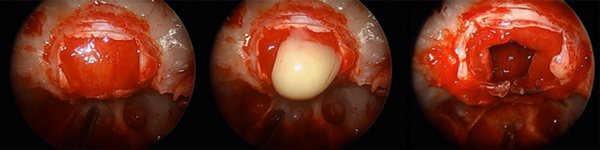
Figura 2. A: Durotomía selar. B: Material purulento. C: Lecho quirúrgico luego de la resección de la lesión.
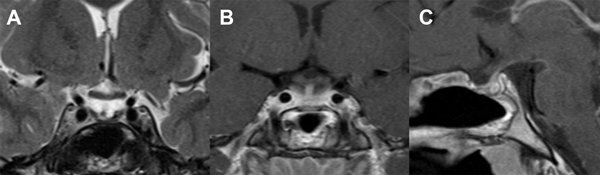
Figura 3. A: IRM T2 coronal. Se visualiza cisterna supraselar libre. B y C: IRM T1 coronal y sagital con gadolinio que muestran hipófisis conservada con captación normal del tallo y glándula.
DISCUSIÓN
Las hipofisitis son un grupo de lesiones inflamatorias que afectan la hipófisis y el tallo hipofisario. La bibliografía actual las divide en primarias y secundarias. Dentro de las primarias se describen varias formas: linfocítica, granulomatosa, xantomatosa, necrotizante, relacionada a IgG4, inducida por inmunoterapia y otras autoinmunes. Las secundarias son aquellas causadas por contigüidad de otra lesión o formando parte de una enfermedad sistémica.
Las hipofisitis primarias son las más frecuentes, y dentro de éstas la forma linfocítica, estimándose 1 caso cada 9 millones de habitantes. También llamada hipofisitis autoinmune, se la distingue según la extensión del proceso inflamatorio en adenohipofisitis, infundibuloneurohipofisitis y panhipofisitis. En un 20-50% de los casos se la asocia a otras entidades autoinmunes: enfermedad tiroidea autoinmunitaria (tiroiditis de Hashimoto, enfermedad de Graves, tiroiditis subaguda), adrenalitis autoinmune, anemia perniciosa y diabetes mellitus tipo 1. Tiene una mayor incidencia en mujeres y en relación con el embarazo y postparto, aunque se han reportado casos en jóvenes y ancianos. Se caracteriza por infiltración difusa de la glándula por linfocitos T y B. La forma de presentación más común comprende disfunción de la adeno y neurohipófisis, cefalea y compromiso visual. Los hallazgos imagenológicos son variables, van desde un agrandamiento de la glándula con realce homogéneo tras el contraste, ausencia de la hiperintensidad en T1 de la neurohipófisis, hasta una silla turca vacía como respuesta atrófica luego del proceso inflamatorio. Con respecto al tratamiento, debe ser adecuado a los signos y síntomas del paciente, siendo posible el manejo conservador, con reemplazo hormonal en caso de ser necesario, ya que es frecuente la resolución espontánea sin disfunción hipofisaria permanente. Los corticoides en altas dosis son de primera línea en casos de cefalea severa y efecto de masa significativo. El uso de inmunosupresores se reserva para casos en que no haya respuesta al tratamiento con corticoides. En cuanto a la cirugía, se indica no solamente para la descompresión quiasmática en casos de resistencia a los corticoides, sino también para la confirmación diagnóstica.
En nuestro caso, la presentación clínica como una meningitis aséptica se asocia más frecuentemente al craneofaringioma, aunque las características de la imagen y la ausencia de disfunción hipofisaria tanto clínica como bioquímica nos llevó a pensar en la posibilidad de un quiste de la bolsa de Rathke. La resección quirúrgica se indicó con el objetivo de obtener el diagnóstico anatomo-patológico y debido a que, si bien el paciente no presentaba déficit visual, en la IRM el tumor se encontraba comprimiendo el quiasma óptico.
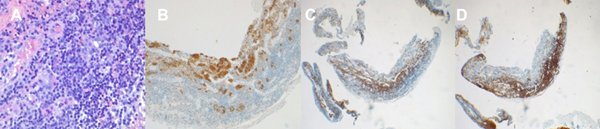
Figura 4. A: Tinción H-E que muestra infiltrado linfoplasmocitario. B: Tinción positiva para sinaptofisina. C: Tinción positiva para CD3 (linfocitos T). D: Tinción positiva para CD20 (linfocitos B).
CONCLUSIÓN
La hipofisitis es una entidad poco frecuente y de difícil diagnóstico clínico ya que no presenta características específicas y puede presentarse de forma muy variable. Debe ser tenida en cuenta como diagnóstico diferencial, sobretodo en pacientes que presentan alguna enfermedad autoinmune. Aunque aún no hay un tratamiento específico, la cirugía endoscópica endonasal es de utilidad para el diagnóstico definitivo y la descompresión de la vía óptica.
BIBLIOGRAFÍA