Del glioma intrínseco difuso de protuberancia al glioma difuso de línea media H3K27M-alterado: ¿cuál ha sido el papel de la biopsia estereotáctica?
Victoria Becerra Castro,1 Mariana Alamar Abril,1 Diego Culebras Palau,1 Santiago Candela Cantó,1 Andrés Morales La Madrid,2 José Hinojosa Mena-Bernal1
1. Servicio de Neurocirugía, Hospital Sant Joan de Déu, Barcelona, España
2. Servicio de Oncología Pediátrica, Hospital Sant Joan de Déu, Barcelona, España
Recibido: 25/05/2024 Aceptado: 05/08/2024
Victoria Becerra Castro
mvictoria.becerra.castro@gmail.com
DOI: 10.59156/revista.v38i03.657
ORCID
Victoria Becerra Castro: 0009-0007-7205-3725
Mariana Alamar Abril: 0000-0002-4942-1717
Diego Culebras Palau: 0009-0007-2102-3323
Santiago Candela Cantó: 0000-0002-7116-7727
Andrés Morales La Madrid: 0000-0001-7083-4316
José Hinojosa Mena-Bernal: 0000-0003-2438-7679
EMAILS
Mariana Alamar Abril: mariana.alamar@sjd.es
Diego Culebras Palau: diego.culebras@sjd.es
Santiago Candela Cantó: santiago.candela@sjd.es
Andrés Morales La Madrid: andres.morales@sjd.es
José Hinojosa Mena-Bernal: jose.hinojosa@sjd.es
Los autores no declaran conflicto de interés
Los autores no declaran financiamiento.
RESUMEN
Introducción: la biopsia estereotáxica en el tumor difuso de protuberancia ha demostrado ser una técnica factible, con alta rentabilidad diagnóstica y baja morbilidad neurológica. Además, ha abierto las puertas a nuevos enfoques terapéuticos basados en la medicina individualizada y los ensayos clínicos, en una enfermedad en la que, actualmente, no existe tratamiento curativo y cuya supervivencia es completamente infausta. Sin embargo, durante años, el papel de este procedimiento quirúrgico en el manejo diagnóstico y terapéutico del paciente pediátrico con glioma pontino intrínseco difuso (GPID) ha sido cuestionado y controvertido.
Objetivos: realizar una revisión histórica sobre la percepción quirúrgica e indicación de la biopsia estereotáctica en el GPID, así como su contribución al conocimiento y comprensión de su compleja biología molecular y, consecuentemente, su repercusión en las nuevas terapias.
Material y métodos: se describen las características generales y de diagnóstico habitual del GPID. Posteriormente, se analizan las limitaciones y consecuencias del diagnóstico clásico, así como su repercusión en el tratamiento de este tipo de tumor.
Resultados: se realiza una viñeta histórica sobre el rol de la biopsia estereotáctica en los tumores de tronco y, en particular, en el GPID, junto con su repercusión en el conocimiento, su biología molecular y avances terapéuticos.
Conclusiones: la biopsia estereotáctica en el tumor difuso de protuberancia es un procedimiento factible con una baja morbi-mortalidad y alta rentabilidad diagnóstica: se aconseja llevarla a cabo en centros médicos experimentados de referencia en neuro-oncología pediátrica.
Palabras clave: Biopsia estereotáxica. Glioma pontino intrínseco difuso. Glioma difuso de línea media H3K27M-alterada. H3K27M.
From diffuse intrinsic pontine glioma to diffuse midline glioma H3K27M-altered: What has been the role of stereotactic biopsy?
SUMMARY
Background: several clinical studies have demonstrated the safety and feasibility of stereotactic biopsy in the setting of Diffuse Intrinsic Pontine Glioma (DIPG) and have contributed to a deeper understanding of the molecular biology of this disease. These developments led to look into in other lines of treatment, based on individualized approach modality and clinical trials, in a disease with no cure and therefore fatal prognosis. However, for many years, the role of the biopsy has been a topic of debate and controversy.
Objective: to perform a historical review about the surgical perception of stereotactic biopsy in DIPG, in addition to contributing to a better knowledge and comprehension of its molecular biology, and consequently, the impact in innovative therapeutic options.
Methods: characteristic clinical symptoms and pathognomonic radiographic features of DIPG are described. Limitations and consequences of classic diagnosis are analyzed, as well as its impact on their treatment.
Results: a historical vignette about the role of stereotactic biopsy in brainstem tumors and in DIPG is elaborated, along with its impact on new developments of molecular pathophysiology for these tumors and novel treatment approaches.
Conclusion: stereotactic biopsy in diffuse intrinsic pontine glioma is a feasible and safe procedure with low morbidity and mortality rate and high diagnostic yield. However, performing DIPG biopsy is recommended to be carried out in experienced and reference medical centers in pediatric neuro-oncology.
Keywords: DIPG. H3K27M. Midline diffuse glioma H3K27M-altered. Stereotactic biopsy.
INTRODUCCIÓN
Gliomas de tronco del encéfalo: focal versus difuso
Glioma de tronco del encéfalo (TE) es un término genérico que comprende un grupo heterogéneo de tumores que se originan en el mesencéfalo, la protuberancia, el bulbo y la médula cervical alta.1 Estos tumores constituyen el 10-20% de los tumores del sistema nervioso central en niños (SNC) y el 25% de los tumores de fosa posterior, con una incidencia aproximada de 250-300 nuevos casos al año en EE. UU.1,2 La media de edad al diagnóstico está entre los 7 a 10 años, sin que presenten una predilección por sexo y sin asociar una herencia genética destacada.3-5
Previo a la implementación de la tomografía y la resonancia, los gliomas de tronco eran considerados y tratados como una entidad patológica única, que asociaban un pronóstico infausto.5 Tal es así que, en 1969, Matson escribía “…brainstem gliomas must be classified as malignant tumors since their location in itself renders them inoperable…”.6 La afirmación de que estos tumores eran malignos por localización y que su resección no era factible fue consecuencia de la ausencia de distinción entre las diferentes partes del TE, a su vez condicionado por técnicas de imagen de baja resolución anatómica (ej., ventriculografía y/o neumoencefalografía) para dicha diferenciación y diagnóstico. Sin embargo, también en esta misma década, se publicaron las primeras series quirúrgicas de la mano de Alvisi y cols. en 1962, Olivecrona y cols. en 1967 y Pool y cols. en 1968.7-10 Olivecrona informó que, de 26 pacientes diagnosticados de tumores localizados en el bulbo, 7 de ellos fueron operados y tuvieron una supervivencia larga entre 10 a 25 años.9 Pool y cols. publicaron 3 casos sometidos a una cirugía con resección parcial y posterior radioterapia, obteniendo una supervivencia de 10 a 22 años.10 Estos resultados alentadores se sumaron tanto a los progresos tecnológicos en el ámbito quirúrgico como al refinamiento de las técnicas de imagen. El advenimiento de la tomografía computarizada en los años 70 y posteriormente de la RM permitió importantes avances en el conocimiento anatómico de estos tumores, ya que facilitó diferenciarlos por localización dentro del tronco del encéfalo y por tipos histológicos. De esta forma, a partir de la década del 80, ligada a la publicación de nuevas series quirúrgicas, se desarrollaron distintos sistemas de clasificación de los gliomas de TE dando por finalizado el concepto del tumor de tronco como entidad única.1,5
Las clasificaciones de los tumores de TE surgieron con el propósito de establecer si el paciente podía beneficiarse de una cirugía y de orientar el pronóstico de la enfermedad. En 1986, Epstein y McCleary clasificaron los gliomas no exofíticos de tronco en tres subgrupos: focal, difuso y cérvico-medular.11 Un año más tarde, Stroink y cols. describieron un nuevo subtipo de glioma de TE con características clínico-radiológicas diferentes a la de los gliomas difusos, que denominaron tumor de TE exofítico dorsal benigno transependimario.12 Posteriormente, el estudio conjunto por parte del Pediatric Oncology Group (POG), el Children´s Hospital of Philadelphia/New York University (CHOP/NYU), y la Universidad de California (UCSF) sobre las resonancias magnéticas de una serie de 87 pacientes con glioma de TE, los llevó a desarrollar una clasificación más detallada sobre la base de los diferentes hallazgos en las imágenes de RM. Los parámetros que tuvieron en cuenta fueron localización, focalidad, dirección del crecimiento, grado de captación y de crecimiento exofítico, presencia o ausencia de quistes, necrosis, hemorragias e hidrocefalia. Se identificaron subgrupos de pacientes en los que se detectó una correlación entre determinadas características de la RM y la supervivencia, entre ellos las lesiones focales (p: 0.005), tumores quísticos (p: 0.03) o con menor captación de contraste (p: 0.05) tuvieron un mejor pronóstico. Asimismo, encontraron una diferencia significativa en la supervivencia en función del sitio primario del origen (p: 0.02). La supervivencia a 2 años fue del 50% para los tumores medulares y del 20% para las lesiones protuberanciales.13 En consonancia con estos resultados, Prados y cols., en 1996, identificaron en su serie de 64 pacientes con gliomas de tronco que el factor más relevante que determinaba el pronóstico según los hallazgos en la resonancia magnética fue si el tumor era difuso o focal. Los tumores focales tenían un pronóstico excelente independientemente de la localización, a diferencia de los tumores difusos, en especial, los pontinos. En los gliomas de protuberancia difusos, la supervivencia al año fue del 46% y a los 3 años del 17%.14
Una de las clasificaciones más recientes y ampliamente aplicadas ha sido la de Choux y cols.; en ella se emplearon las imágenes de TC y RM para clasificar a los tumores de TE en cuatro grupos: I, tumor intrínseco, difuso; II, tumor focal e intrínseco, puede ser sólido o quístico; III, exofítico, lateral o dorsal; y IV, cérvico-medular. El objetivo no ha sido otro que facilitar la elección del plan terapéutico. Para los tumores del grupo I, la piedra angular de tratamiento era la radioterapia, su comportamiento intrínseco-difuso no hacía factible una cirugía resectiva, a diferencia de los otros tipos tumorales donde la cirugía tenía un papel clave inicial.15 Posteriormente, Guillamo y cols., en 2001, y Sousa y cols., en 2004, clasificaron los tumores de TE en base a la modalidad terapéutica de la que son subsidiarios. De forma que los tumores focales, exofíticos y bulbomedulares fueron considerados inicialmente candidatos de un tratamiento quirúrgico, en contraposición de los tumores difusos, cuyo tratamiento lo constituye fundamentalmente la radioterapia. También, hacen distinción de los tumores tectales, una entidad clínico-radiológica de excelente pronóstico, su curso indolente, a diferencia de los anteriores, los hace candidatos de una conducta expectante.16,17
Así pues, en el análisis retrospectivo de todos estos sistemas de clasificación subyace una distinción dicotómica entre tumores de TE focales frente a difusos; mientras que en estos últimos la resección quirúrgica no juega ningún papel, la extirpación tumoral puede ser una opción de tratamiento en los tumores focales. A partir de ahí, el mayor o menor grado de complejidad de dichas clasificaciones sobre la base de los hallazgos de la RM no es otro que intentar predecir de forma más detallada el comportamiento biológico del tumor y, por consiguiente, la expectativa pronóstica, así como el manejo terapéutico más apropiado.1
OBJETIVOS
Realizar una revisión histórica sobre la percepción quirúrgica e indicación de la biopsia estereotáctica en el GPID, así como su contribución al conocimiento y comprensión de su compleja biología molecular y, consecuentemente, su repercusión en las nuevas terapias.
MATERIAL Y MÉTODOS
Características generales del tumor difuso intrínseco de protuberancia o glioma pontino intrínseco difuso (GPID)
El GPID constituye el 10-15% de los tumores pediátricos y el 75-80% de los tumores de tronco del encéfalo. Son lesiones difusas que comprometen la protuberancia, su evolución clínica es agresiva y su pronóstico infausto.18 La edad al diagnóstico está comprendida entre los 5 y 10 años. Los síntomas iniciales son variables, pero de relativa rápida instauración (pocas semanas) y resultan de la afectación de estructuras fundamentales del TE. Al diagnóstico, los pacientes pueden presentar la tríada clásica de síntomas consistente en la afectación de pares craneales (diplopía, parálisis facial), disfunción de las vías largas y afectación cerebelosa (ataxia, disartria y dismetría), aunque no siempre se acompaña de todos los déficits neurológicos. El aumento de la presión intracraneal e hidrocefalia obstructiva por el efecto expansivo del tumor en la protuberancia es infrecuente al diagnóstico, pero puede ocurrir durante la evolución de la enfermedad.18,19
La media de supervivencia global de los pacientes con GPID oscila entre los 8 a 13 meses y la supervivencia libre de progresión es de 5 a 8 meses. Además, la supervivencia a los 2 y 5 años es inferior al 10% y al 1%, respectivamente,18,20-22 convirtiéndose en una de las principales causas de muerte en niños con tumores cerebrales.18,23
Diagnóstico clásico del GPID: limitaciones y consecuencias
Históricamente, el diagnóstico del GPID es clínico y radiológico. Se fundamenta en una sintomatología de rápida evolución con afectación de pares craneales y/o signos de vías largas, unido a unos hallazgos radiológicos clásicos definidos por una masa expansiva difusa con al menos el 50% de la protuberancia afectada. Es un tumor que se caracteriza por ser hipointenso en T1 e hiperintenso en T2/FLAIR, con alguna zona de captación de contraste (aunque no siempre) de morfología anular, rodeando a áreas de necrosis o inflamación. Por lo que, conforme estos criterios relativamente específicos a esta entidad, muchos pacientes han sido y son tratados sin una confirmación histológica.23 Además, su localización profunda e infiltrante impide una extirpación quirúrgica total, y en ocasiones ni siquiera parcial. De hecho, en las décadas de los 80 y 90, tras intentarlo, esta conducta terapéutica fue abandonada porque no mejoraba el pronóstico del paciente, con el riesgo añadido de generar secuelas considerables.24
Por otra parte, los hallazgos histológicos de autopsias y el curso clínico agresivo condujeron a la idea errónea que la biología del GPID era similar a la del glioblastoma del adulto o a la de otros gliomas malignos supratentoriales pediátricos.25-28 Según estas creencias se extrapolaron regímenes de tratamiento de gliomas de alto grado del adulto a niños con GPID que, sin sorprender, no lograron frenar o retrasar significativamente la progresión de estos tumores.20 Tal es así, que la combinación de radioterapia y temozolomida, tratamiento médico de primera línea en el glioblastoma del adulto, no mejoraron la supervivencia en el GPID pediátrico.20,29
Actualmente, la radioterapia normofraccionada con una dosis de 54-60 Gy durante 6 semanas es el tratamiento estándar en los pacientes con GPID, si bien su eficacia es transitoria.30 Este tratamiento proporciona una mejoría o control temporal de los síntomas, reduce la necesidad de esteroides y prolonga la supervivencia en un promedio de 3 meses, mientras la media de supervivencia es de 4.5 cuando no se realiza la radioterapia.18,31 Otras modalidades de radioterapia como la terapia hiperfraccionada (dosis más pequeñas y frecuentes) no mejoró la supervivencia global y conllevó un aumento de la toxicidad,32-35 en contraposición a la radioterapia hipofraccionada (dosis más elevadas durante un período de 3 semanas) que, si bien no mejoraba el pronóstico, sí demostró que era igual de “eficaz”, tenía un impacto positivo en la calidad de vida y acortaba el tiempo de hospitalización en pacientes muy sintomáticos.36,37
Posteriormente, la radioterapia se ha combinado con otros tratamientos con el objetivo de mejorar el pronóstico funcional y la supervivencia. No obstante, durante muchos años, el avance en el desarrollo de terapias dirigidas se vio obstaculizado primero, por la falta de conocimiento en la biología molecular de estos tumores (por ausencia de muestras tumorales) y segundo, y consecuencia de lo anterior, por el concepto erróneo de que la caracterización genética y molecular era similar a sus contrapartes adultas o a otros tumores gliales de alto grado supratentoriales pediátricos.23,38
RESULTADOS
Compresión histórica sobre el rol de la biopsia estereotáctica en las lesiones del tronco del encéfalo y en especial, en el GPID
El estudio de la biología molecular del GPID se ha visto limitado por la falta de tejido tumoral disponible. Debido a la localización dentro de la protuberancia, durante mucho tiempo se pensó que realizar biopsias de estas lesiones se asociaba con una morbilidad inadmisible en pacientes que ya tenían una corta esperanza de vida.11,39 La biopsia estereotáctica de los tumores de TE es un procedimiento antiguo, el cual fue impulsado después de los primeros informes sobre la seguridad y eficacia de biopsias en lesiones cerebrales profundas en adultos publicados en la década del 70.40,41 En 1985, Coffey y Lunsford describieron las vías de abordaje para la realización de la biopsia estereotáctica según la localización de las lesiones en TE con buenos resultados quirúrgicos y, posteriormente, en esta misma década continuaron las publicaciones en las que se incluían tanto pacientes adultos como pediátricos que documentaban un buen rendimiento diagnóstico con una baja tasa de complicaciones.42-46 Sin embargo, a medida que mejoró la resolución de la resonancia magnética, el papel de la biopsia se volvió controvertido para los pacientes con tumores infiltrantes difusos para quienes las opciones de tratamiento seguían estando limitadas a la radioterapia. Tal es así que, en 1990, Allbright, del Children´s Cancer Group afirmó “... la resonancia magnética brinda imágenes que son prácticamente diagnósticas de gliomas de tronco encefálico y brindan información pronóstica equivalente a la que se obtiene de las biopsias...”.47
Décadas después, continuó siendo un tema controvertido con actitudes contrapuestas según los grupos quirúrgicos, unos renunciaron a realizarla argumentando los riesgos potenciales y la ausencia de un beneficio directo para el paciente, y otros continuaron indicándola fundamentalmente en el contexto de lesiones o imágenes atípicas o dentro de ensayos clínicos.48
Curiosamente, Hakinson y cols., en 2011, publicaron los resultados de una encuesta sobre la evaluación por parte de los neurocirujanos pediátricos de los hallazgos de resonancia magnética de ejemplos seleccionados de GPID típico versus atípico. Los autores concluyeron, que “…la práctica de diagnosticar GPID en función de las características de imagen y la historia clínica por sí sola no alcanza el umbral adecuado para ser considerada un estándar de atención…”.49 Posteriormente, Sufit y cols. informaron de una serie de biopsias de 7 pacientes con tumores difusos de protuberancia. Dos de los siete fueron DNET, tras lo cual concluyeron: “…Dado que el diagnóstico por imágenes no es confiable y la biología de los tumores es dispar, se debe realizar una biopsia para permitir un diagnóstico preciso y dirigir tratamientos potencialmente más efectivos…”.50
A partir de la década del 2000 se publicaron múltiples series sobre biopsias estereotáxicas en lesiones en el tronco del encéfalo, lo cual proporcionó un conocimiento significativo y valioso. Pero muchos de estos artículos estuvieron constituidos por series mixtas de pacientes adultos y pediátricos con diagnósticos muy variables. En dichos artículos, la tasa de morbilidad estuvo entre un 0 a un 25% y la de mortalidad entre un 0 a un 3%. Sin embargo, cuando se ahonda extrayendo los datos de los pacientes pediátricos con GPID, se puso de manifiesto una morbilidad inferior al 5% en las series más largas sin mortalidad y una rentabilidad diagnóstica entre el 96 y el 100%.48
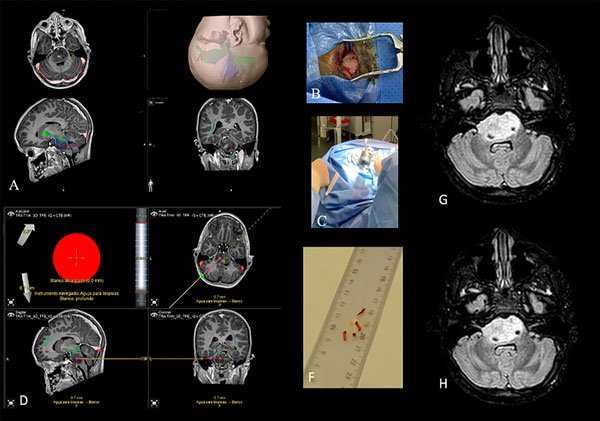
El metaanálisis que realizó Samadani y cols. en 2004 incluyó 13 estudios con un total de 293 biopsias de TE en adultos y niños. La morbilidad asociada al procedimiento fue del 4% de forma transitoria, del 1% de manera permanente y se produjo un fallecimiento en un paciente adulto con una lesión vascular (mortalidad, 0.3%). La rentabilidad diagnóstica fue del 96%.51 Posteriormente, en el 2006, Pincus y cols. publicaron su serie sobre biopsia de lesiones de tronco del encéfalo en pacientes pediátricos, así como una amplia revisión de la literatura sobre esta técnica puramente en niños. En un total de 192 pacientes incluidos por los diferentes estudios se obtuvo una rentabilidad diagnóstica del 94.9%, una tasa de morbilidad del 4.9% y de mortalidad del 0.7%.52 En 2010, Rajshekhar publicó una serie de 106 con tumores de tronco del encéfalo biopsiados en la que consiguió una rentabilidad diagnóstica del 100% y tuvo lugar una morbilidad transitoria del 4%, sin mortalidad.53 Posteriormente, la siguiente serie larga publicada sobre biopsias de TE y específicamente en GPID fue la del grupo del Hospital Necker (París) en 2015. En ella, se recogieron 130 procedimientos con una tasa de déficit neurológico transitorio del 3.9%, sin mortalidad ni secuelas permanentes y una rentabilidad diagnóstica del 100%.48 En esta línea de estudio, cabe señalar que, en una extensa serie de 270 casos de adultos sometidos a una biopsia estereotáctica, la biopsia realizada sobre lesiones pontinas no constituyó un factor de riesgo de morbilidad significativa.54 Esto se confirma con los estudios anteriores mencionados, así como por otros más recientes. En 2017, en el metaanálisis realizado por Hamisch y cols. en el cual revisaron un total de 735 pacientes con tumores de TE pediátricos que se sometieron a una biopsia estereotáxica obtuvieron una tasa de rentabilidad diagnóstica del 96.1%, una morbilidad global del 6.7%, aunque permanente de solo 0.6% y una mortalidad del 0.6%.55 De igual forma, diferentes series unicéntricas publicadas por varios grupos como el de la Universidad de California (San Francisco, EE. UU.), el Children's Hospital of Michigan (Detroit, EE. UU.), el Bambino Gesù Children’s (Roma, Italia), Great Ormond Street Hospital (Londres, Reino Unido) y dos trabajos multicéntricos han demostrado que las biopsias en GPID se pueden obtener de forma factible y segura con una alta rentabilidad diagnóstica en centros médicos experimentados.56-61 El tejido tumoral obtenido por las biopsias estereotáxicas unido a los estudios de autopsias ha proporcionado una compresión más profunda sobre la biología molecular y genética del GPID. En la Figura 1 se ilustra el procedimiento quirúrgico de una biopsia estereotáxica en un GPID.
Figura 1. A) Planificación preoperatoria con la trayectoria para la realización de la biopsia estereotáctica en un GPID. B-D) Abordaje quirúrgico. F) Cilindros de biopsia tumoral. G-H) Imágenes de control postquirúrgico.
En las últimas décadas, los avances constantes en el campo de la neurooncología molecular ha permitido identificar alteraciones que son potencialmente susceptibles de beneficiarse de terapias dirigidas, si bien estas solo pueden determinarse mediante perfiles moleculares personalizados a través de tejido que se obtiene mediante una biopsia tumoral. Este concepto terapéutico también ha sido implementado en el GPID. La idea inicial de renunciar a la biopsia previo comenzar el tratamiento (radioterapia) debido a la ubicación profunda del tumor y a las opciones limitadas de tratamiento ha ido cambiando progresivamente. Por un lado, de la mano del progreso en los análisis moleculares y genéticos también lo ha hecho la mentalidad de realizar una biopsia en el momento del diagnóstico no solo para confirmarlo sino para obtener las características biológicas del tumor. Y, por otro lado, la limitación técnica en el pasado para realizar una trayectoria segura al tumor62 ha sido solventada por las tecnologías de neuronavegación y las modalidades de imágenes de alta resolución, como se explicó anteriormente.19,63 Además, otra de las ventajas que brinda la biopsia es una posible inclusión en ensayos clínicos que precisan la confirmación de cierta alteración. Desde el punto de vista de la investigación, el tejido tumoral resulta valioso para establecer líneas celulares tumorales que permitan probar y validar nuevos tratamientos en cultivos celulares o modelos animales.38
DISCUSIÓN
Del GPID al glioma difuso de línea media (DMG) H3K27M-alterada
En el trabajo de Jansen y cols., de 108 biopsias realizadas en 13 estudios, se halló que el GPID englobaba un conjunto variable de grados histológicos tumorales, entre los cuales se describieron 20 astrocitomas difusos grado II de la Organización Mundial de la Salud (OMS), 1 oligodendroglioma grado II, 1 oligoastrocitoma grado II, 37 astrocitomas anaplásicos grado III, 3 oligoastrocitoma grado III, 27 glioblastomas multiforme, 15 gliomas malignos y 4 tumores histológicamente indefinidos.20 Tanto en este informe como en la serie de Necker,48 a diferencia de los adultos, el grado de tumor no se correlacionó con el comportamiento tumoral ni la supervivencia global o supervivencia libre de progresión.64 Un artículo sobre casos de autopsias reveló unos hallazgos similares, confirmando que el esquema tradicional de clasificación de la OMS por grados no tiene capacidad predictiva en el GPID.65
Posteriormente, en el 2018, el estudio colaborativo de los registros de GPID de la Sociedad Internacional y Europea de Oncología Pediátrica (SIOP) informó que, de los 299 pacientes biopsiados y de las 77 necropsias, la variación histológica clásica fue también similar a los estudios anteriores, y no encontraron diferencias significativas entre los distintos grados y la supervivencia global,66 es decir, los pacientes con una lesión de bajo grado evolucionaban igual de mal que aquellos con una histología tradicional de alto grado.66,67
Tanto esta divergencia entre las lesiones del adulto y del paciente pediátrico como el mal pronóstico independientemente de la histología clásica viene explicada por la distinta firma molecular de los tumores. Los estudios de biopsias demostraron que el GPID es una entidad tumoral biológicamente diferente a la del glioblastoma del adulto. Las mutaciones que caracterizan estos tumores de alto grado del adulto, dentro de las cuales se incluyen algunas como el IDH1, FGFR1, MYB y TERT son atípicas en el GPID.65,68 En el 2012 se publicaron varios estudios relevantes que identificaron la mutación de la histona 3 (H3) K27M en casi el 80% de los casos de GPID.69-71 Posteriormente, también se detectó la mutación de H3K27M hallada en el GPID en otros gliomas de localización talámica y medular, lo cual era sugestivo de una alteración de la regulación epigenética central subyacente a dicha “oncohistona”.72-74 Es por ello que esa mutación fue considerada reveladora en términos de la comprensión fisiopatológica de dichos tumores, y llevó, en 2016, a la reclasificación de los tumores del sistema nervioso central (SNC) de la OMS con una nueva entidad denominada “glioma de línea media difusa H3K27M-mutado”, siendo considerados como un grado IV de la OMS independientemente de los criterios histológicos convencionales.75
El panorama epigenético y de expresión genética completo del GPID es muy complejo, extenso y en ciertos aspectos todavía desconocido. Uno de los mecanismos moleculares claves para la compresión de la patogénesis del GPID fue el descubrimiento de que la mayoría de ellos (80%) albergaban una mutación determinada y recurrente, la H3K27M, en uno de los dos genes que codificaban las histonas, las cuales son estructuras claves de la cromatina que desempeñan un papel importante en la regulación del epigenoma.63 Esta mutación se puede dividir en dos alteraciones genéticas específicas. Por un lado, está la H3.3 en el gen H3F3A detectada en el 65-70% de los casos, y cuya función en condiciones normales se relaciona con el reemplazo de histonas según sea necesario en caso de interrupción del nucleosoma. Y, por el otro lado, se encuentra la mutación H3.1 en el gen HIST1H3B y, menos frecuentemente, en el HIST1H3C. Su actividad está relacionada con el empaquetamiento del ADN recién sintetizado en la fase S.21 En ambas variantes, la alteración detectada fue una mutación “missense” –sin sentido– específica en la que se producía una sustitución de la lisina por la metionina en la posición 27 (K27M) ubicada en la cola N-terminal de la histona.70,71 Así pues, estamos ante una mutación de ganancia de función con efectos transcripcionales al eliminar la trimetilación H3K27M. La consecuencia es una represión de los genes diana del complejo represivo 2 de Polycomb (PRC2) a través de la interacción del K27M mutado con la subunidad EZH2, conduciendo a una desrepresión aberrante y robusta de la transcripción génica con desagregación de la cromatina y aneuploidía celular normalmente silenciada por PRC2.19
La mutación H3K27M es heterocigota en el 100% de las células de GPID y, lo que es más destacado, es que permanece así tanto en zonas de alto grado como de bajo grado, y en muestras expuestas como en aquellas no tratadas.21 Pese a que el papel completamente exacto de la mutación H3K27M en la tumorogénesis no está completamente dilucidado,76,77 sí está demostrado que la mutación es un promotortumoral y su presenciase asocia a un peor pronóstico.78 En un metaanálisis realizado por Mackay y cols. en 2017 obtuvieron que la presencia de las mutaciones K27M tanto en H3.3 como en H3.1 se asociaron de forma independiente con una supervivencia más corta (p <0.0001).79 Además, se ha encontrado una correlación entre la clínica y el fenotipo histológico-molecular en los GPID H3K27M-mutado. Se ha informado que aquellos pacientes que albergaban la mutación en H3.1 tuvieron un inicio más temprano de la enfermedad (aprox. 2 años, p<0.0001) y lo que es más relevante, presentaron una mejor respuesta clínica a la radioterapia (85% versus a 55.3% de buenas respuestas clínicas, p = 0.0263) y una supervivencia global algo más prolongada (mediada de 15.0 meses versus 9.2 meses, p = 4.51e-05). Además, estas mutaciones fueron impulsoras de dos programas oncogénicos distintos, en los tumores H3.3 K27M (gen H3F3A) se objetivó un fenotipo proneural-glioblastoma, oligodendrocítico o neural junto con una regulación negativa significativa en los genes inhibidos en las metástasis, que se tradujo en una mayor tasa de recaídas metastásicas en el análisis comparativo (p = 0.04). Por el contrario, en los tumores con la mutación H3.1 se identificó la sobreexpresión de genes relacionados con un fenotipo de glioblastoma mesenquimatoso y células astrogliales, unido a regulación positiva de genes implicados en la angiogénesis y la hipoxia con una activación de HIF1A. Estas características moleculares tuvieron su representación en la resonancia con zonas extensas necróticas y de edema extracelular mayor que en los tumores H3.3. Tal es así, que dichas mutaciones exhibieron diferente expresión radiológica en la difusión. Los valores de ADC y DDC fueron en general más bajos y la extensión de la distribución menor en los tumores H3.1 en comparación con los H3.3 (ADC: 1228 y 1612 µm 2/s, p <0.001 y DDC: 1499 y 2688 µm 2/s, p <0.001).80
Entre los gliomas de línea media H3K27M-mutado la supervivencia también se vio condicionada por la localización anatómica. En 2018, Karreman y cols. objetivaron que los pacientes con gliomas de línea media talámica H3K27M tuvieron una supervivencia más prolongada que aquellos con GPID o tumores medulares y, claramente, menor en las tres ubicaciones que sus contrapartes sin la mutación.78
En las últimas décadas, la comprensión científica del perfil molecular del GPID se ha incrementado sustancialmente y, si bien la mutación de la histona 3 (H3) K27M fue considerada patognomónica, se han identificado otras alteraciones genéticas, individualmente poco frecuentes, pero que pueden representar subgrupos moleculares específicos.63 En el 2014, tras la secuenciación completa de veinte GPID, dio como resultado a una clasificación en tres subgrupos moleculares: MYCN, “silent” y H3K27M. Todos ellos con unas características clínicas, genéticas y epigenéticas particulares, aunque no se objetivó una diferencia significativa en la supervivencia entre estos subgrupos.65 En esta línea, se han ido detectando otras aberraciones genéticas entre las que se incluyen mutaciones en el complejo ATRX, que suele tener una alta presencia en tumores H3K7M mutado;69,79 alteraciones de componentes dentro de la vía RTK-RAS-PI3K observadas hasta en el 40-68% de los GPID y habitualmente en el grupo H3K27M mutado, un ejemplo son amplificaciones o mutaciones de ganancia de función de PDGFRA (36-50%),81,82 mutaciones FGFR o aumento de MET, EGFR y ERBB1.27,83,84 Otra alteración detectada hasta en un 25% de los pacientes con GPID en los análisis retrospectivos fue la mutación en el receptor de activina A tipo 1 (ACVR1).84,85 Se ha identificado su concurrencia con la mutación H3K27M, estando las dos presentes hasta en un 22% de los GPID y la mayoría asociados a la variante H3.1.84,85 La presencia de la mutación ACVR1 también se asoció a una edad más temprana en la aparición del tumor (mediana de edad 5 años) y una supervivencia global discretamente más prolongada.86 Otras alteraciones identificadas han sido la ganancia de los componentes de la vía RTK y RB, incluidos KRAS, AKT1, PIK3CA, CDK4 y CDK5 (47-69%).27,71
La mutación en la proteína tumoral p53 (TP53) hallada hasta en un 75% de los casos de GPID y con mayor coocurrencia con la mutación H3K27M.19,87 En un estudio retrospectivo, los GPID con H3K27M mutada y TP53 tuvieron una mayor resistencia a la radioterapia, mayor agresividad tumoral y peor supervivencia global en comparación con los pacientes sin ninguna de las alteraciones o con una sola de ellas.87 Aunque probablemente la mutación H3K27M constituyó el impulsor oncogénico más relevante, la presencia de ambas mutaciones conllevó peor evolución clínica, lo que sugirió un efecto sinérgico que asociaba un fenotipo más agresivo.19 Otro hallazgo fue la sobrexpresión y amplificación del factor transcripcional MYCN y su proteína diana PVT1. El subgrupo molecular MYCN del GPID se caracterizó por la hipermetilación del ADN y el reordenamiento cromosómico, que genera aneuploidía. Se piensa que la vía MYCN puede estar inducida por la mutación H3K27M, pero que, por lo demás, constituyen vías completamente independientes.19,88 Otras afectaciones identificadas fueron las mutaciones en la vía WNT, que conllevan un aumento de los niveles de ß-catenina, proteína clave en la señalización de WTN, que induce la proliferación celular.88
También, se describió la sobreexpresión del gen que codificaba el factor de transcripción del desarrollo PAX3 (40%) y la desregulación de la vía Hedgehog (Hh).89 Asimismo, se ha publicado que las mutaciones de ganancia de función en ACVR1 tuvieron lugar en tumores en la protuberancia con H3.1 K27M en contraposición con las mutaciones/fusiones de FGFR1 que ocurrían en tumores talámicos asociados a H3.3 K27M. Las alteraciones de ganancia de función en tres receptores del factor de crecimiento: ACRV1, FGFR1 y PDGFRA se asociaron con variantes H3 K27M en los gliomas de línea media, si bien no se objetivaron al mismo tiempo y además las mutaciones ACRV1 y FGFR1 fueron mutualmente excluyentes con las alteraciones TP53 y conducen localizaciones específicas dentro de la línea media encefálica.90
Dentro de este panorama molecular tan complejo en los gliomas difusos de línea media, se ha informado de unas alteraciones genéticas también muy relevantes aparte de la conocida mutación K27M. Estas fueron la sobreexpresión de la proteína EZHIP y alteraciones en el EGFR. La primera, la sobreexpresión EZHIP, fue observada por primera vez en los ependimomas tipo A y lleva a una reducción global de H3K27Mme3. En el trabajo de Castel y cols. se observó que 9 de 241 casos (3.7%) tenían una histología clásica de GPID y pérdida de la trimetilación de H3K27M, pero que carecían de positividad para K27M por inmunohistoquímica. Cuando analizaron la expresión de EZHIP vieron su sobreexpresión sistemática, lo que los llevó a plantear que el espectro de glioma difuso de línea media (GDM) con inhibición de PRC2 iba más allá de la mutación H3K27M, proponiendo un nuevo subgrupo de tumores EZHIP/H3-WT que podrían considerarse similares a los gliomas de línea media H3K27M mutada.91
En esta misma línea, Mondal y cols. describieron la existencia de otro subconjunto de gliomas difusos, de localización fundamentalmente talámica o bitalámica, que tenían una amplificación y/o mutación del EGFR y pérdida de H3K27me3. La pérdida de la trimetilación parecía estar mediada tanto por la mutación H3K27M como por la sobreexpresión de EZHIP. Los autores concluían que la pérdida de H3K27me3 debería considerarse como un rasgo común de tres clases moleculares diferentes de glioma difuso de línea media pediátrico: el glioma difuso de la línea media “típico” con la mutación H3K27M, el glioma difuso de la línea media con sobreexpresión de EZHIP (que además mostraba una alta frecuencia de mutaciones AC‐ VR1), y los gliomas difusos principalmente bitalámicos que presentaban sobreexpresión de H3K27M o EZHIP junto con un fuerte enriquecimiento para alteraciones de EGFR.92,93
La importante trascendencia biológica de todos estos descubrimientos condujo en el año 2021, en la quinta edición de la clasificación de tumores del SNC de la OMS, a una nueva designación de “glioma difuso de línea media, H3K27M-altered”. El objetivo fue tener en cuenta aquellos tumores con un mecanismo alternativo para la pérdida de la trimetilación H3K27M (sobreexpresión de GDM EZHIP, GDM EGFR mutado), además de la más común que es la mutación H3K27M. De tal forma que en la nueva subclasificación se integran los siguientes subtipos:94
Tras la identificación de las múltiples mutaciones en el GPID y la mayor comprensión de la naturaleza heterogénea de estas lesiones, han surgido ensayos clínicos que han combinado la biopsia estereotáctica con terapias dirigidas a determinadas alteraciones moleculares. Así pues, en el estudio de “DIPG Biology and Treatment Study” se evaluó la viabilidad de la biopsia en el momento del diagnóstico para el análisis genético y estratificación de grupos de tratamiento, de manera que los pacientes inscriptos podían recibir terapias aprobadas por la FDA en función de las mutaciones halladas. De forma que los pacientes fueron tratados con radioterapia y bevacizumab, pero también podían recibir erlotinib en función de la presencia de la mutación EGFR y temozolomida en caso de la metilación del promotor MGMT. En este estudio no se informó mortalidad y la morbilidad fue mínima con una hemiparesia en un paciente. La rentabilidad diagnóstica fue elevada, consiguiendo tejido suficiente para estudios genéticos y moleculares en el 96% de los procedimientos y, también, demostró que la biopsia no interfería con el período de tiempo clínicamente apropiado para comenzar con la radioterapia (media de inicio: 10 días, rango: 4-22 días).60 En esta misma línea, el ensayo clínico internacional “Biological Medicine for Diffuse Intrinsic Pontine Gliomas Eradication (BIOMEDE)” tuvo como objetivo principal comparar la eficacia de tres fármacos (dasatinib, everolimus, erlotinib) combinados con la radioterapia en términos de supervivencia global. Dichos fármacos fueron asignados según el perfil molecular del tumor (PTEN-pérdida, EGFR-sobreexpresión) que fue definido gracias a la realización de una biopsia estereotáxica al diagnóstico de GPID. No se informó mortalidad asociada a la biopsia, la rentabilidad diagnóstica fue elevada con solo 5 biopsias no concluyentes y señalaron que dicho procedimiento descartó el diagnóstico de GPID en un 8% de los casos. Este estudio puso de manifiesto la viabilidad del tratamiento dirigido en el GPID y condujo a un segundo ensayo clínico según el mejor perfil de toxicidad y eficacia ligeramente mejor (aunque no estadísticamente significativa) del everolimus.95
CONCLUSIONES
La indicación de la biopsia estereotáctica en el GPID ha sido un tema controvertido, y en ocasiones desestimado por la ubicación del tumor y las pobres expectativas terapéuticas. Sin embargo, conforme ha aumentado el conocimiento sobre su biología molecular, también lo ha hecho la mentalidad de plantear una biopsia en el momento del diagnóstico, abriendo las puertas a opciones innovadoras de tratamiento en el contexto de ensayos clínicos o terapias personalizada. Si bien múltiples grupos han demostrado que la biopsia es un procedimiento factible con una baja morbi-mortalidad y alta rentabilidad diagnóstica, se aconseja su realización en centros médicos experimentados de referencia en neurooncología pediátrica.
Este es un artículo de acceso abierto bajo la licencia CC BY-NC https://creativecommons.org/licenses/by-nc/4.0/
BIBLIOGRAFÍA