Síndrome de regresión caudal: reporte de un caso
Dayana Ariza Echavarria,1 Carlos Del Toro Trillos,2 Daniela Paez Jimenez,3 Juan Salcedo Brand,4
1. Servicio de Radiología e Imágenes Diagnósticas, Sabbag Radiólogos, Barranquilla, Colombia.
2. Unidad de Cuidados Intensivos, Clínica Bonadonna, Barranquilla, Colombia.
3. Servicio Hospitalario, Clínica La Asunción, Barranquilla, Colombia.
4. Servicio de Radiología e Imágenes Diagnósticas, Hospital Universidad del Norte, Barranquilla, Colombia.
Recibido: octubre 2023. Aceptado: enero 2024.
Carlos Del Toro
carlosdt1106@gmail.com
DOI:10.59156/revista.v38i01.559
ORCID:
Dayana Ariza Echavarría 0000-0002-0000-1607
Carlos Del Toro Trillos 0000-0003-2377-7050
Daniela Páez Jiménez 0000-0001-7892-2257
Juan Salcedo Brand 0000-0001-7222-4044
RESUMEN
Introducción. El síndrome de Regresión Caudal es una malformación congénita poco frecuente que se caracteriza por la afectación musculoesquelética caudal y que puede estar asociado a defectos neurológicos, espinales, gastrointestinales, renales y genitourinarios.
Objetivos. Presentar el caso de una paciente con síndrome de Regresión Caudal y discutir sus características clínicas, el diagnóstico y manejo multidisciplinario.
Descripción del caso. Paciente de 2 años con antecedentes de diabetes gestacional tratada con insulina que consulta por infecciones urinarias a repetición. Ecografía renal con dilatación pielo-calicial y TC que evidencia ausencia de cuerpos sacro-coxígeos.
Intervención. La paciente recibió un manejo multidisciplinario no quirúrgico y fue dada de alta para seguimiento ambulatorio.
Conclusión. El síndrome de regresión caudal debe ser pensado en paciente con antecedentes de diabetes gestacional e infecciones urinarias en los primeros años, destacando la importancia del oportuno diagnóstico prenatal. El manejo multidisciplinario es vital para optimizar la evolución de estos pacientes.
Palabras clave. Anomalías congénitas. Diabetes gestacional. Malformación lumbosacra. Regresión caudal.
Caudal Regression Syndrome: Case Report
ABSTRACT
Background. The Caudal Regression Syndrome is a rare congenital malformation characterized by caudal musculoskeletal involvement and which may be associated with neurological, gastrointestinal, renal and genitourinary defects.
Objectives. To describe a patient with Caudal Regression syndrome and to discuss its clinical characteristics, diagnosis and multidisciplinary management.
Description of the case. A 2-year-old patient with a history of being born after gestational diabetes treated with insulin, presented with urinary tract infections. Renal ultrasound with pyelocalyceal dilation and CT scan showing absence of sacrococcygeal bone bodies.
Intervention. The patient received non-surgical management with a multidisciplinary team and was discharged for outpatient follow-up.
Conclusion. Caudal regression syndrome should be considered in patients with a history of gestational diabetes and urinary infections in the early years, highlighting the importance of timely prenatal diagnosis. Multidisciplinary management is vital to optimize the evolution of these patients.
Keywords. Caudal regression. Congenital anomalies. Gestational diabetes. Lumbosacral malformation.
INTRODUCCIÓN
El síndrome de Regresión Caudal, también conocido como síndrome de agenesia sacra o displasia caudal, es una malformación congénita poco frecuente que se caracteriza por la afectación musculoesquelética caudal y que puede estar asociado a defectos neurológicos, gastrointestinales, renales y genitourinarios. El síndrome puede detectarse como un hallazgo incidental o puede presentarse con agenesia sacra o lumbosacra con afectación de los segmentos de médula espinal y con alguna malformación musculoesquelética de la pelvis y extremidades inferiores.1 Aunque no existen datos consistentes sobre la incidencia global del síndrome, se ha descrito un espectro de malformaciones que van desde el ano imperforado hasta la sirenomelia.2
La etiología del síndrome no es clara aún, pero se considera multifactorial, con una predisposición genética y la exposición a factores como la diabetes materna. Se cree que el defecto podría iniciarse antes de la cuarta semana de gestación, en la inducción de los primordios caudales del embrión.1,3
El objetivo del reporte es presentar el caso clínico de una paciente con síndrome de Regresión Caudal y discutir sus características clínicas, el diagnóstico y manejo multidisciplinario.
DESCRIPCIÓN DEL CASO
Se trata de una paciente de 2 años de edad, segunda gestación, madre de 39 años, con diabetes gestacional tratada con insulina. A los 13 meses fue diagnosticada con desnutrición aguda, estreñimiento y ectasia urétero-pielo-calicial bilateral asociada a infecciones urinarias a repetición.
La paciente consultó con un cuadro clínico de 2 días de evolución, caracterizado por dolor en región lumbar izquierda con irradiación hacia hemiabdomen inferior, sintomatología irritativa urinaria y síndrome febril. Al ingreso, se encontró taquicárdica y con hipertensión arterial, así como con talla/edad, peso/edad, peso/talla y perímetro cefálico/edad por debajo de -2 desviaciones estándar.
En los laboratorios se observó leucocitosis por aumento del recuento de neutrófilos, Proteína C reactiva elevada, injuria renal aguda KDIGO II, acidemia metabólica compensada y ecografía de abdomen total reportando dilatación bilateral del tracto urinario, asociada a vejiga de esfuerzo, dilatación y engrosamiento de asas del intestino delgado, y moderada ascitis ecogénica. Se lo interpretó como un abdomen agudo secundario a sospecha de infección de vías urinarias, injuria renal aguda vs crónica agudizada y ectasia pielo-calicial bilateral. Se inició manejo con antibióticos empíricos y líquidos endovenosos de mantenimiento; al persistir la paciente con dolor abdominal de gran intensidad y timpanismo, fue evaluada por cirugía pediátrica, descartando un abdomen agudo quirúrgico y solicitando una tomografía de abdomen y pelvis.
INTERVENCIÓN
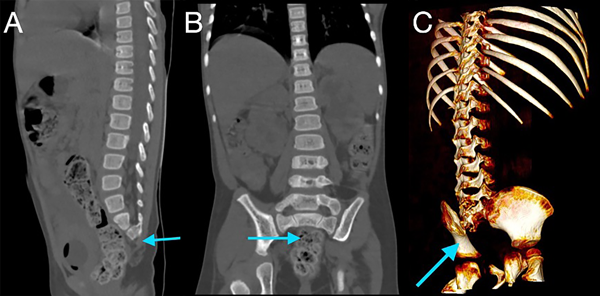
En la tomografía se evidenció presencia de S1-S2 con ausencia del resto de cuerpos sacro-coxígeos, así como hipodensidad a nivel de la médula que podría sugerir siringomielia (Figura 1). También se encontró falta de unión de algunos elementos posteriores a nivel de L5-S1 (Figura 2), lo que sugiere el diagnóstico de síndrome de regresión caudal asociado a probable siringomielia y probable vejiga neurogénica dado engrosamiento marcado de las paredes vesicales (Figura 3). Además, se encontró dilatación ureteropielocalicial de forma bilateral (Figura 4). A raíz de estos hallazgos, se solicitó una valoración por genética clínica, la cual determinó que la paciente cumple con los criterios clínicos de regresión caudal y tiene antecedentes de diabetes gestacional insulino-dependiente, lo cual es un factor de riesgo.
Figura 1. Tomografía de abdomen simple en ventana ósea, reconstrucciones sagital (A), coronal (B) y 3D (C), mostrando la presencia de vertebras sacras 1 y 2, con ausencia del restante de los elementos óseos sacro-coxígeos (flechas azules).
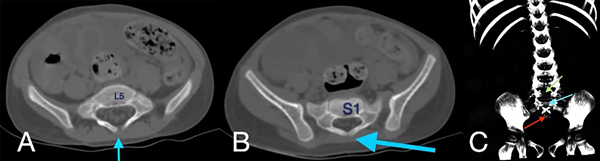
Figura 2. Tomografía de abdomen en ventana ósea, cortes axiales (A, B) y reconstrucción en 3D (C), donde se evidencia ausencia de unión de los elementos posteriores de L5 (flecha azul en A, flecha verde en C) y S1 (flecha azul en B y C) y ausencia de elementos sacro-coccígeos (flecha roja en C).

Figura 3. Tomografía de abdomen, cortes axiales, donde se muestra dilatación de la pelvis renal derecha (A), e izquierda (B), así como de los trayectos ureterales hasta distal y discreto engrosamiento de sus paredes (C, flecha azul derecho, flecha roja izquierdo).
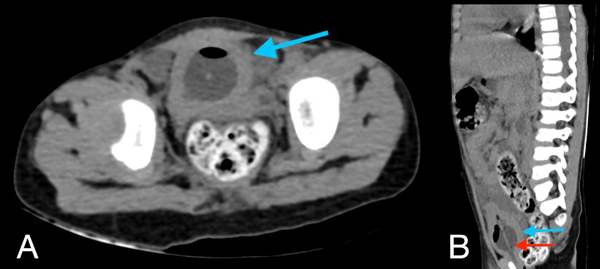
Figura 4. Tomografía de abdomen en corte axial (A) y sagital (B): la vejiga evidencia engrosamiento concéntrico de sus paredes de aproximadamente 7,4 mm (flecha azul en A y B), con la presencia de balón de sonda Foley en su interior (flecha roja en B).
Después de estabilizar las cifras de presión arterial, completar el tratamiento antibiótico y no existiendo conducta quirúrgica a tomar, se decidió el alta hospitalaria de la paciente. Se indicó continuar con el manejo de la hipertensión arterial mediante antagonistas cálcicos vía oral cada 12 horas y se programó seguimiento ambulatorio por parte de los especialistas en Pediatría, Nefrología, Urología, Genética y Nutrición.
DISCUSIÓN
El síndrome de Regresión caudal (SRC) o displasia caudal es una rara malformación congénita que se presenta en 0,1-0,25 casos por cada 10.000 embarazos normales y con una prevalencia de 1:10.000 nacidos vivos. Los reportes indican que los hijos de madres diabéticas tienen una incidencia 200 veces mayor que la población general, con 1 de cada 350 casos presentando SRC.4 Descripto por primera vez en 1852, el SRC es un espectro de malformaciones que pueden variar desde la existencia de defectos aislados en el desarrollo del sacro, con ausencia parcial o total de éste, hasta el grado más grave conocido como “sirenomelia”. Esta afección se origina por un fallo primario en el mesodermo durante el desarrollo embrionario, que impide la correcta inducción de un número adecuado de somitas caudales. Esto da lugar a la fusión de los primordios de los miembros y a la falta de formación -o desarrollo incompleto- de las estructuras caudales.2
Pang y cols. clasificó el SRC en 5 tipos según la gravedad y la extensión de la malformación: Tipo I y II agenesia total del sacro con y sin agenesia vertebral lumbar; tipo III agenesia sacra parcial con preservación de al menos S1; tipo IV hemisacro; y tipo V agenesia del cóccix.5
El diagnóstico de SRC puede realizarse en etapa prenatal por medio de la ultrasonografía, siendo los casos diagnosticados en el primer trimestre los más graves y letales, como es el caso de sirenomelia. Lo habitual es que el diagnóstico se realice en el segundo trimestre ya que se puede visualizar con mayor detalle anatómico, permitiendo detectar la agenesia sacro-coccígea y, además, la valoración del resto de las malformaciones asociadas.2
La tomografía computarizada de la columna vertebral es una herramienta útil para detectar anomalías óseas, ya que permite identificar los segmentos afectados, la forma y tamaño de los cuerpos vertebrales y la posición de la pelvis.6 Por otro lado, la resonancia magnética es especialmente importante para evaluar el compromiso del tubo neural y las lesiones intra y extradurales. Esto permite diferenciar entre los dos grupos más frecuentes de la clasificación de Pang: el tipo I (41%), donde el cono termina por encima del nivel normal y se asocia con un canal central dilatado; y el tipo II (59%), donde el cono medular está alargado y se encuentra caudalmente anclado, por debajo de L1.5,6
El SRC se asocia con diversos trastornos, incluyendo malformaciones gastrointestinales, genitourinarias, esqueléticas y otros tipos de malformaciones. Entre las malformaciones genitourinarias, se pueden encontrar genitales ambiguos, hidronefrosis, pielectasia, dilatación de la vejiga, agenesia renal unilateral o bilateral, riñón fusionado o en herradura y uréter(es) ectópico(s).7
En este caso, se encontró una ausencia parcial sacro coxígea con preservación de S1 (Figura 1), asociada a no unión de elementos posteriores de L5 y S1 (Figura 2), siendo un síndrome de regresión caudal tipo I según Pang, asociado a alteraciones genitourinarias dadas por hidronefrosis bilateral (Figura 3) y engrosamiento de las paredes vesicales (Figura 4).
CONCLUSIÓN
En resumen, el SRC es una malformación congénita compleja y poco común que puede presentarse con diferentes grados de gravedad, afectando no sólo la región sacro coccígea, sino también otros sistemas del cuerpo, como el tracto urinario. La detección temprana del síndrome es esencial para comprender mejor su extensión y gravedad, y un diagnóstico precoz puede ayudar a implementar un tratamiento adecuado y reducir los riesgos de complicaciones. Por lo tanto, es fundamental realizar una evaluación clínica detallada junto con la interpretación de imágenes radiológicas para evaluar el grado de afectación de los segmentos vertebrales, lo que permitirá una intervención terapéutica efectiva y adecuada.
Los autores no declaran conflicto de interés
Los autores no declaran financiamiento.
Este es un artículo de acceso abierto bajo la licencia CC BY-NC https://creativecommons.org/licenses/by-nc/4.0/
BIBLIOGRAFIA